Precision Genome Annotation, Redefined.
Unmatched Accuracy. Customized Insights. Proven Expertise.










































Driven by Customer Impact
High-quality annotations delivered on time, powering real-world breakthroughs.
Stay Ahead with
Weekly Bioinformatics Insights
GOENOMICS delivers cutting-edge genome annotation solutions powered by breakthrough research. Serving industry leaders in plant and animal breeding, biotechnology, and academia, we specialize in high-precision annotations for plant, animal, and fungal genomes.
Our proprietary mendle® technology sets a new standard by dramatically reducing false positives, ensuring highly accurate and reliable genome annotations for your research and innovation success.
Products
Genome annotation service
Annotation of all genes - protein-coding, RNA and more
Genome annotation means that each individual nucleotide in a genome sequence is assigned a function. Our technology is based on more than 35 years of academic research and experience. Genome annotation is the basis for many costly experiments. So start your research with the best data you can get and let us annotate the genome for you.
We annotate
protein-coding and non-coding genes, RNA genes, transposons and pseudogenes
We assign
UTR regions, coverage and information about biological function
We generate
comprehensive data reports based on biological information
Get deep biological insights through enhanced analysis reports
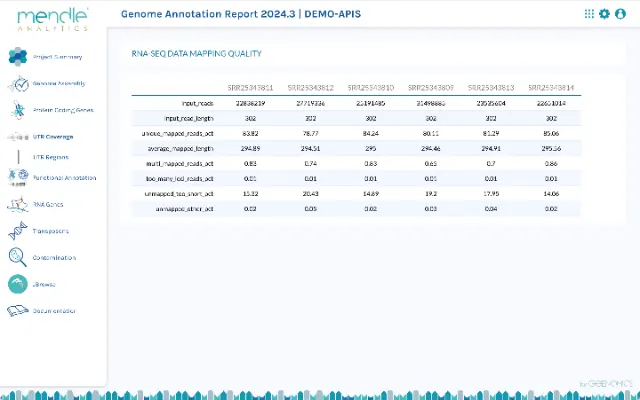
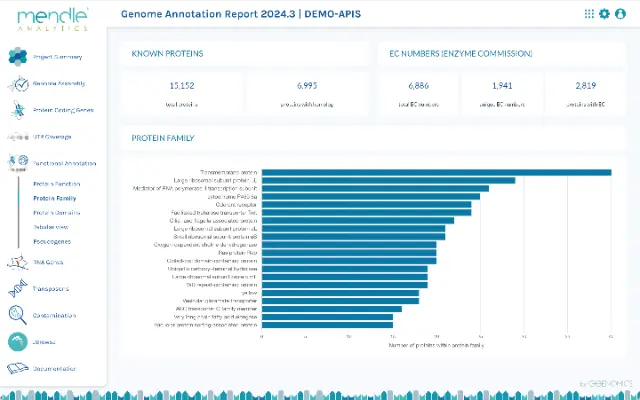
Genome annotation is the beginning of biological analyses. To give you the best possible start, we create a detailed analysis report for each annotation with diagrams, tables and information about the genome structure and each analyzed gene type. The genes are related to biological functions, e.g. the decoding capacity of the tRNA genes in the genetic code, the completeness of spliceosome complexes or the expansion of certain protein families.
Cloud based report
Immediate access to results
Bioinformatics experience not required


What our customers say
Scientist
Sartorius Stedim Cellca, Germany
Dr. Virginia Friedrichs (Researcher)
Institute of Diagnostic Virology, Friedrich-Loeffler-Institut
Federal Research Institute for Animal Health
Researcher
Department of Botany and Plant Science, University of California Riverside
USDA-ARS National Germplasm Repository for Citrus & Dates
Your gateway to genomic discovery
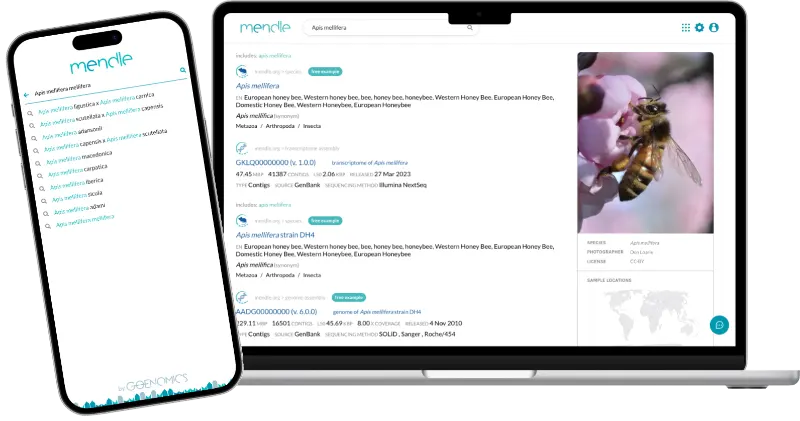
Search platform for genome assemblies and annotations
Knowledge about already sequenced genomes is distributed across many institutional databases (e.g. GenBank, Ensembl, CNGBdb), university-hosted species/taxon databases and individual data repositories (e.g. figshare, dryad, github). Mendle unites all genome assembly data worldwide in a single entry platform! Search results are linked to genome assembly and annotation analyses.
Looking for Verticillium longisporum strains for which genome assemblies are available?
Founding team

Dr. Martin Kollmar
CEO and Founder

Dr. Dominic Simm
CTO and Co-Founder
Trusted connection
GOENOMICS and Dovetail Genomics announce strategic partnership
Partnership to provide faster, richer annotation services for evolutionary biology, conservation, and agriculture
Supported by


